
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Proinflammatory cytokine IL-17A identified as a key factor in non-alcoholic steatohepatitis, a condition that could lead to hepatocellular carcinoma
Blocking IL-17A could prevent this condition in patients at risk, in particular with diabetes or hepatitis viral infection
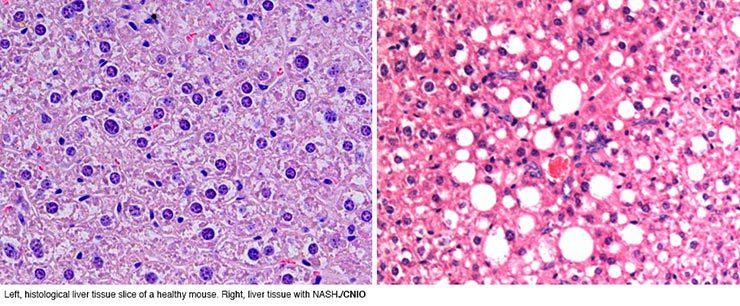
Non-alcoholic steatohepatitis (NASH) is a serious hepatic condition that precedes hepatocellular carcinoma (HCC) and is currently untreatable. A study conducted at the Spanish National Cancer Research Centre (CNIO) shows that a proinflammatory molecule, IL-17A, is a key factor in the development of this pathology, and points out that blocking IL-17A or inhibiting cells that secrete IL-17A with drugs such as digoxin (an antiarrhythmic agent) may be useful to prevent NASH in patients susceptible to develop HCC.
HCC is the most aggressive liver neoplasm and a major cause of cancer-related morbidity. Several risk factors have been associated with this carcinoma and its early stages, but the molecular mechanisms underlying the carcinogenic process remain unclear.
Non-alcoholic fatty liver disease (NAFLD), characterized by excessive lipid accumulation, is highly prevalent among obese individuals, viral-infected patients or people with diabetes, and it’s an important risk factor for HCC development. However, not all obese individuals evolve to the most severe form, NASH that has an important inflammatory component.
“Accumulation of fat (steatosis) by itself cannot explain the appearance of NASH. On the contrary, inflammation determines the progression and outcome of the disease, as only 10% to 20% of obese patients with fatty liver disease will eventually develop NASH”, explains Nabil Djouder, leader of the study. What Djouder and his colleagues have observed is that NASH, which is currently untreatable, is the result of several “hits” and that “the first step is DNA damage promoting inflammation caused by the excess of nutrients”.
Working with different mouse models, the authors demonstrated in the latest issue of Cancer Cell how excess of nutrients stimulate the expression of an oncogene called URI in the liver. URI –which is also up-regulated in viral hepatitis- leads to DNA damage in the hepatocytes and this triggers a systemic inflammation and a cross-talk between white adipose tissue and liver that ultimately ends up in NASH.
When DNA damage appears in the hepatocytes, immune cells infiltrate the liver, especially Th17 cells that release the proinflammatory molecule IL-17A. This molecule, which is a cytokine, induces neutrophil infiltration of the adipose tissue that leads to insulin resistance and fatty acid release, resulting in NASH. “Type 2 diabetes seems to precede NASH and HCC”, said Djouder.
The researchers also treated healthy mice with IL-17A injections and observed how the first signs of NASH appeared after four weeks, confirming its crucial role in the disease development. Moreover, Djouder and his team blocked IL-17A using various methods –antibodies and digoxin among others- and prevented the development of NASH and HCC.
Also, URI expression and IL-17A levels were positively associated with non-alcoholic stetatohepatitis and HCC in obese, HVB and HCV patients. This finding should pave the way for a new prevention strategy for NASH and HCC in patients at high risks, in particular with diabetes or hepatitis viral infection.
“Treatment of HCV represents a socio-economic challenge for our society in that blocking IL-17A […] with digoxin may provide effective and inexpensive prophylaxis for hepatitis B and C infected patients with high NASH and HCC risks”, concludes the paper.
This research project has been funded by the Spanish Ministry of Economy and Competitiveness, the European Foundation for the Study of Diabetes (EFSD) and Worldwide Cancer Research (WCR).
Reference article
Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Ana L. Gomes, Ana Teijeiro, Stefan Burén, Krishna S. Tummala, Mahmut Yilmaz, Ari Waisman, Jean-Philippe Theurillat, Cristian Perna, Nabil Djouder. Cancer Cell (2016). DOI: http://dx.doi.org/10.1016/j.ccell.2016.05.020